





FDA 510(k)
According to different risk levels, the US Food and Drug Administration (FDA) divides the medical devices into three classes (I, II, and III), with Class III medical devices having the highest risk level.

If you plan to enter the US market, in addition to the quality management system that needs to comply with the US GMP requirements (commonly referred to as QSR820), you also need to go through the establishment registration and product listing. Among them, the vast majority of Class I medical devices can be directly listed, and a very small number of products are exempt from GMP; for most Class II medical devices, as well as a small number of Class I and Class III medical devices, a 510(k) application needs to be submitted to the FDA to obtain the clearance, and then the products can be listed; for the remaining Class III medical devices, a PMA (Premarket Application) needs to be submitted to the FDA to obtain the clearance, and then the products can be listed.
1. What is 510(k)
FDA510(k) is the pre-market notification (PMN), which aims to prove that the product is substantially equivalent to the product that has been legally marketed. It is commonly referred to as 510(k) because it is described in Section 510(k) of the Food, Drug, and Cosmetic Act (FD&C Act). For most Class II devices, as well as a small number of Class I and Class III devices, a 510(k) application needs to be submitted to the FDA before they are marketed.
2. Under what circumstances shall the FDA510(k) be submitted
· A medical device is introduced to the US market for the first time
· The intended use of a device already on the market is changed
· A device that has been marketed is changed or updated (such change or update will affect the safety or effectiveness of the device, involving the design, material, chemical composition, driving force, production process or intended use)
3. Who must apply for FDA510(k)
Section 510(k) of the FD&C Act does not specify who must apply for 510(k) - anyone can apply. However, it specifies the kind of behavior for which a 510(k) application is required, such as introducing a device to the US market. Based on the specified behavior, the following persons must submit the 510(k) application to the FDA:
1) Manufacturers which introduce devices to the US market;
2) R&D designers which introduce devices to the US market;
3) Repackagers which change the devices or device labels;
4) Foreign manufacturers/exporters or US agents of foreign manufacturers/exporters which introduce devices to the US market
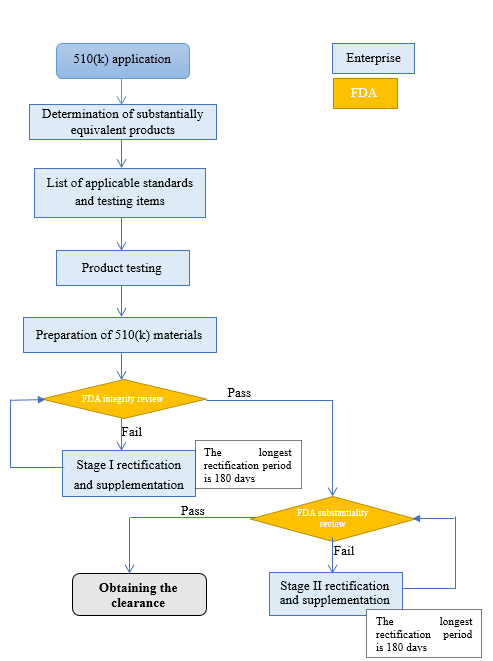
4. 510(k) application process
5. As a global technical service provider that is professional in the field of medical device regulations, SUNGO can provide the following services:
1)Determine the specific classification and risk level of your medical device in the US;
2)Counsel the enterprise on preparing the materials required for the 510(k) application;
3)Determine the testing standards and applicable items, recommend the testing institutions, and review the testing reports;
4)Prepare the 510(k) declaration documents;
5)Counsel the enterprise on applying for the small business qualification;
6)Track the 510(k) document review progress;
7)Counsel the enterprise on the rectification and supplementation;
8)Perform establishment registration and product listing.
6. SUNGO service process
Process | Task | Division of work | Cycle |
Stage I: 510K declaration | |||
1 | Overall understanding; development of the testing protocol Including searching and determining the comparison device, and listing the information and testing items needed for document preparation | Sungo takes the charge The enterprise assists in the confirmation | 15 working days |
2 | The enterprise prepares the testing samples according to the testing requirements, and contacts the laboratory for testing price inquiry and sample submission for testing | The enterprise makes the preparation Sungo provides the guidance | Subject to the testing time |
3 | Preparation of the materials listed for the 510(k) application | Enterprise | 1-2 months |
4 | Application for the small business qualification | The enterprise files the application Sungo provides the guidance | 1-2 months |
5 | 510k document preparation | Sungo | 15~30 working days |
6 | 510K document information confirmation | Enterprise | 1 week |
7 | Payment of the FDA review fee | Enterprise | 1 week |
8 | Document submission for review | Sungo | 3~5 working days |
9 | Format review | FDA | 7days after receiving the document |
10 | Rectification and communication | Sungo | 1~2 working days |
11 | Integrity review | FDA | 15 days after receiving the document |
12 | Rectification and communication | By Sungo and the enterprise *The longest rectification period is 6 months | 3~5 working days |
13 | Substantiality review | FDA | Within 60 days after receiving the document |
14 | Rectification and communication | By Sungo and the enterprise *The longest rectification period is 6 months | 15~20 working days (If additional tests are required, the actual testing time shall prevail) |
15 | The review is over and the K number is approved | FDA | Within 90 days after receiving the document |
Stage II: establishment registration & product listing | |||
16 | PIN application | Sungo | 2 working days |
17 | Payment of the annual fee in USD based on the PIN | Enterprise | 7 working days |
18 | Completion of establishment registration & listing | Sungo | 5 working days |
