





Product & Production Recording
I. Main legal basis
Regulations for the Supervision and Management of Medical Devices
Measures for the Supervision and Administration of Medical Device Production
Measures for the Administration of Registration of Medical Devices
Announcement of the China Food and Drug Administration on Matters Concerning the Recording of Class I Medical Devices
Good Manufacturing Practice for Medical Devices
II. Recording declaration
Certificate type:
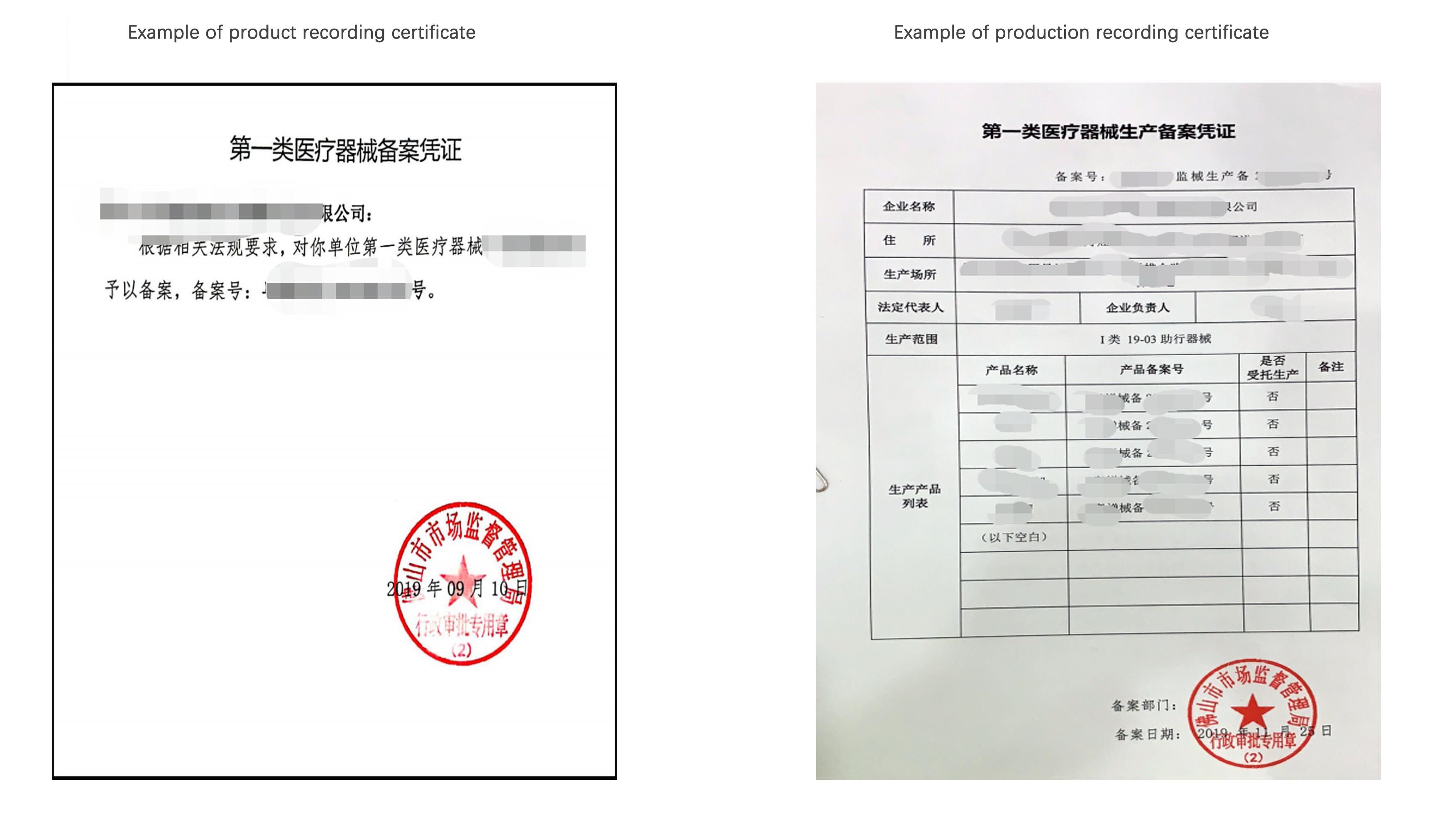
Product recording certificate & production recording certificate
For domestic Class I medical device products, the product recording certificate and the production recording certificate are both required before they can be formally produced and sold in China.
Recording process:
For the recording of the Class I medical device products, the recorders shall submit the recording materials to the local municipal food and drug administration departments of the cities with subordinate districts.
The product inspection reports thereof may be the self-inspection reports of the recorders, and the clinical evaluation materials thereof exclude the clinical trial reports, and may be the materials capable of proving the safety and effectiveness of the medical devices through the literatures or the data obtained from the clinical application of similar products. In addition, for an imported Class I medical device, a representative office established or a corporate legal person designated by the overseas manufacturer in China shall act as an agent, providing the food and drug administration departments of the State Council with the recording materials as well as the documents proving that the competent authorities of the country (region) of the recorder approve the medical device to be marketed.
The production recording declaration shall be made after completing the product recording. Generally, the production recording materials shall be submitted to the local district-level food and drug administration department of the district with subordinate districts.
System assessment:
The enterprise that applies for the recording of Class I medical devices shall establish and improve the quality management system compatible with the medical devices it produces and ensure the effective operation of the system in accordance with the requirements of the Good Manufacturing Practice for Medical Devices in combination with the product characteristics.
The inspection content involved in the GMP quality management system mainly includes the followings:
(I) Organization and personnel
(II) Plants and facilities
(III) Equipment
(IV) Document management
(V) Design and development
(VI) Purchasing
(VII) Production management
(VIII) Quality control
(IX) Sales and after-sales service
(X) Non-conforming product control
(XI) Adverse event monitoring, analysis and improvement
Certificate maintenance and update:
The product recording certificate and production recording certificate are valid permanently.
When the matters stated in the recording materials change, the recording shall be changed in the original recording department.
III. Declaration documents
The product recording application materials generally include the followings:
(I) Product recording application form;
(II) Product risk analysis document;
(III) Product technical requirements;
(IV) Product inspection report;
(V) Clinical evaluation document;
(VI) Manufacturing information;
(VII) Samples of Instructions for Use and labels;
(VIII) Supporting documents;
(IX) Declaration of conformity;
(X) Letter of authorization.
The production recording application materials generally include the followings:
(I) Class I medical device production recording form;
(II) Medical device recording certificate of the product;
(III) Product technical requirements that have been recorded;
(IV) Business license;
(V) ID cards of the legal representative and the person in charge of the enterprise;
(VI) Proof of the identities, academic qualifications and titles of the persons in charge of production, quality and technology;
(VII) List of the employees on the production management and quality inspection positions and their academic qualifications and titles;
(VIII) Documentary evidence of production site (lease contract/property certificate);
(IX) Catalogue of main production equipment and testing equipment;
(X) Quality manuals and procedure documents;
(XI) Process flow charts;
(XII) Self-guarantee statement for the authenticity of the application materials;
(XIII) Letter of authorization for the handler.
IV. Service content
SUNGO can provide services related to the Chinese regulations, including:
· Develop the application solutions
· Make the medical device classification definition declaration
· Assist the enterprise to establish the GMP or GSP management system
· Prepare the medical device recording/registration dossiers
· Assist the enterprise to obtain the product recording/production recording certificates
· Assist the enterprise to obtain the product registration certificate/production license/operation license
· Assist in obtaining the product recording/registration certificates of imported medical devices
· Provide the training and counseling on medical device regulations
· Renewal of registration/change of licensed items/change of registered items
· Production license renewal/production license change
V. Service process
Process | Specific task | Division of work | Cycle |
1 | The enterprise provides the preliminary product information, and SUNGO determines the product classification and the declaration path | Both parties | 2-3 working days |
2 | SUNGO signs the cooperation agreement with the enterprise | Both parties | 2-3 working days |
3 | SUNGO assists the enterprise to establish a quality management system that meets the GMP | Both parties; SUNGO provides the counseling in the whole course | 3-6 months |
4 | Product recording declaration | Both parties; with the support from the enterprise, SUNGO prepares the materials and completes the declaration | 1-3 months |
5 | Recording review by NMPA, and rectification | Both parties; SUNGO guides the completion of the rectification declaration | 1-2 months |
6 | Issuance of product recording certificate | NMPA | 1 working day |
7 | Production recording declaration | Both parties; with the support from the enterprise, SUNGO prepares the materials and completes the declaration | 5-10 working days |
8 | Issuance of production recording certificate | NMPA | 1 working day |
9 | System inspection and rectification | Both parties; with the support from the enterprise, SUNGO provides guidance for receiving the review | 2 working days |
10 | After the end of the process, Class I medical devices can be produced | N/A | N/A |
